ReaxFF molecular dynamics simulations of CO collisions on an O-preadsorbed silica surface
ReaxFF molecular dynamics simulations of CO collisions on an O-preadsorbed silica surface
P. Gamallo, H.Prats, R.Sayós
J. Mol. Model. 20 (2014) 2160.
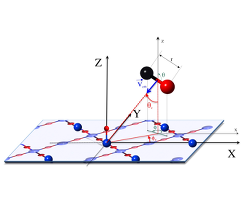
CO molecule interacting with an O-preadsorbed β-cristobalite (001) surface
A quasiclassical trajectory dynamics study was performed for carbon monoxide collisions over an oxygen preadsorbed β-cristobalite (001) surface. A reactive molecular force field(ReaxFF) was used to model the potential energy surface. The collisions were performed fixing several initial conditions: CO rovibrational states (v = 0-5 and j = 0, 20, 35), collision energies (0.05 ≤ E col ≤ 2.5 eV), incident angles (θ v = 0°, 45°) and surface temperatures (T surf = 300 K, 900 K). The principal elementary processes were the molecular reflection and the non-dissociative molecular adsorption. CO 2 molecules were also formed in minor extension via an Eley-Rideal reaction although some of them were finally retained on the surface. The scattered CO molecules tend to be translationally colder and internally hotter (rotationally and vibrationally) than the initialones. The present study supports that CO + O ad reaction should be less important than O + O adreaction over silica for similar initial conditions of reactants, in agreement with experimental data.