Kinetic Monte Carlo simulations of the water gas shift reaction on Cu(111) from density functional theory based calculations
Kinetic Monte Carlo simulations of the water gas shift reaction on Cu(111) from density functional theory based calculations
H.Prats, L. Álvarez, F. Illas, R.Sayós
Journal of Catalysis (2015) (In press)
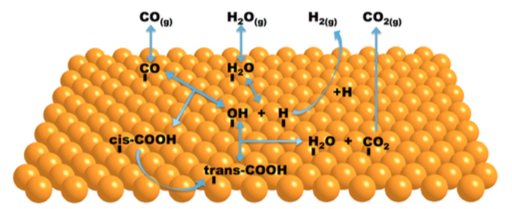
A systematic first-principles kinetic Monte Carlo study of the water gas shift reaction taking place on the Cu(111) surface is presented including adsorption/desorption, diffusion and other elementary chemical reactions, totalling 34 elementary steps with all reaction rates obtained from periodic density functional theory based calculations. The kinetic Monte Carlo simulations were carried out at different partial pressures and temperatures. The results show that the diffusion processes cannot be neglected and that the reaction proceeds predominantly through an associative mechanism via a carboxyl intermediate. The analysis of temperature dependence shows an Arrhenius behaviour with an apparent activation energy of 0.5-0.8 eV in agreement with experiments and with previous microkinetic studies. The effect of H2O/CO ratio on this reaction shows that mixtures with higher CO proportion enhance the reactivity, also in accordance to previous studies. The present work allows one to ascertain the relative importance of the different steps in the mechanism of water gas shift reaction over Cu(111) at several conditions as well as to see the coverage evolution of the surface.