First-principles study of structural, elastic and electronic properties of α-, β- and γ -graphyne
First-principles study of structural, elastic and electronic properties of α-, β- and γ -graphyne
A. Ruiz-Puigdollers, P. Gamallo.
Carbon 96 (2016) 879.
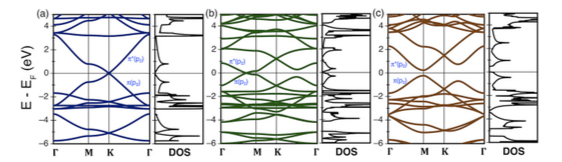
Band structure and total density of states for (a) α-. (b) β-, and (c) γ-graphyne.
This study presents different properties of 2D carbon allotropes: α-, β- and γ-graphynes based on Density Functional Theory calculations. Among the set of properties studied there are structural (e.g., cohesive energies, optimum lattice parameters, interatomic distances, pore sizes, planar packing densities and specific surface areas), mechanical (e.g., uniaxial along with homogeneous and heterogeneous biaxial strains, in-plane stiffness and Poisson’s ratio) and electronic properties (e.g., band structures, total and projected density of states, Dirac points or band gap location, Fermi velocity associated to the formers and the effective masses of the carriers associated to the latter). Basically, graphynes present lower values than graphene for cohesive energy, planar packing density, in-plane stiffness and Fermi velocities. Contrarily, graphynes posses a higher variety of pore sizes and a higher Poisson’s ratio. These facts make graphynes potential candidates for a wide variety of membrane separations and for applications that need softer materials but maintaining, in a lower extent, some of the electronic properties considered exceptional in graphene.