Assessing the activity of Ni clusters supported on TiC(001) toward CO2 and H2 dissociation.
P. Lozano-Reis, H. Prats, R. Sayós, J. A. Rodriguez, F. Illas.
J. Phys. Chem. C 125 (2021) 12019-12027.
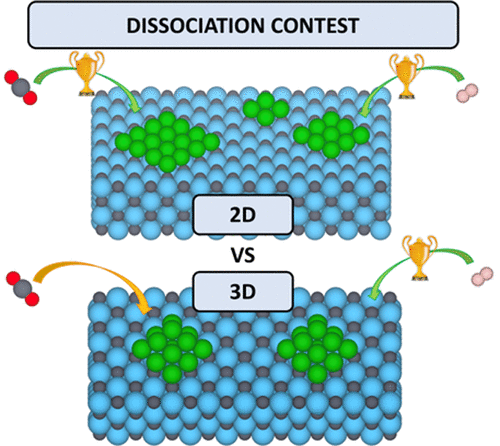
Small Ni particles supported on TiC(001) were shown to display a very high activity for the catalytic hydrogenation of CO2 but the underlying chemistry is, to a large extent, unknown. Here, by means of periodic density functional theory (DFT) calculations with the BEEF–vdW functional, we explore the adsorption and subsequent dissociation of CO2 and H2 on several Nin clusters (n = 4, 9, 13, and 16) supported on TiC(001) and compare the results to those obtained for the bare Ni(111) and TiC(001) surfaces using exactly the same computational approach. The calculations reveal that the Nin/TiC system exhibits stronger adsorption energies and lower dissociation energy barriers for CO2 and H2 than the bare Ni(111) and TiC(001) surfaces. This is in line with the experimental finding evidencing that the Ni/TiC system has a catalytic activity higher than that of the separated Ni and TiC constituents. In addition, the calculated results show that two-dimensional (2D) supported clusters adsorb CO2 and H2 stronger than the three-dimensional (3D) supported clusters and also the 2D clusters exhibit lower energy barriers for CO2 dissociation. Within the 2D supported clusters, larger particles feature slightly stronger adsorption energies and lower CO2 dissociation energy barriers. Finally, H2 dissociation proceeds with a very low energy barrier on all of the studied models, which makes these novel systems potential good candidates for hydrogenation reactions.